

用k-mer分析进行基因组调查(genome survey) —— 用GenomeScope评估基因组特征+用Smudge...
source link: https://yanzhongsino.github.io/2022/06/05/omics_genome.survey_GenomeScope/
Go to the source link to view the article. You can view the picture content, updated content and better typesetting reading experience. If the link is broken, please click the button below to view the snapshot at that time.
用k-mer分析进行基因组调查(genome survey) —— 用GenomeScope评估基因组特征+用Smudgeplot估计倍性
2022-06-05omics , genome , genome survey
【推荐】用Smudgeplot评估物种倍性后,用组合jellyfish+GenomeScope1.0做二倍体物种的基因组调查,用组合KMC+GenomeScope2.0做多倍体物种的基因组调查。
1. k-mer进行基因组调查的软件概况
k-mer进行基因组调查分为k-mer频数统计和基因组特征评估两步。
- GenomeScope可以实现第二步基因组特征评估。
- 需要在jellyfish/KMC等软件的第一步结果k-mer频数分布表的基础上,GenomeScope才可实现。
推荐第一步获取k-mer频数分布表的命令
jellyfish
jellyfish count -C -m 21 -s 1000000000 -t 10 *.fastq -o sample.jf #计算k-mer频率,生成sample.jf
jellyfish histo -t 10 sample.jf > sample.histo #生成k-mer频数直方表sample.histo和k-mer直方图-
mkdir tmp
ls *.fastq.gz > FILES
kmc -k21 -t16 -m64 -ci1 -cs10000 @FILES kmcdb tmp #计算k-mer频率
kmc_tools transform kmcdb histogram sample.histo -cx10000 #生成k-mer直方图
2. GenomeScope概况
- GenomeScope可以利用第一步jellyfish或KMC等其他软件分析得到k-mer频数分布表(sample.histo文件)实现第二步基因组特征评估。
- GenomeScope1.0在2017年发表,用于二倍体物种的基因组调查;2020年又发表了GenomeScope 2.0版本,用于多倍体物种的基因组调查,并发布了用于判断物种倍性的Smudgeplot。
- GenomeScope1.0的基因组特征结果包括基因组大小(genome size),杂合度(heterozygosity),重复序列比例,GC含量等;GenomeScope2.0的基因组特征结果包括基因组大小(genome size),杂合度(heterozygosity),重复序列比例,GC含量,基因型比例,和基因组结构(同源/异源多倍体)等。
- GenomeScope有网页版和Linux本地版,功能一样;推荐网页版,免去安装的麻烦。
3. GenomeScope1.0 网页版【推荐】—— 适用于二倍体物种
GenomeScope1.0 网页版:http://qb.cshl.edu/genomescope
- 上传第一步获得的k-mer频数分布表sample.histo文件;
- 设置参数Kmer length为第一步选择的k-mer长度值,这里是17;
- 参数Read length为序列读长,一般为150;
- 参数Max kmer coverage默认是1000。建议按照物种情况修改,比如10000,以统计更准确。这个参数太小,可能造成过滤过多的Kmer,导致估计的基因组大小偏小的情况。这个参数太大则可能把高拷贝数量的DNA,比如叶绿体DNA,包括进Kmer的统计,造成GenomeScope算法的误差,所以还是不推荐使用-1或太大的值。
- 提交后几分钟就可以得到结果,保存结果图片可用于发表。

Figure 1. GenomeScope1.0结果示例
4. GenomeScope2.0 网页版 【推荐】 —— 适用于多倍体物种
GenomeScope2.0 网页版:http://qb.cshl.edu/genomescope/genomescope2.0
GenomeScope2.0版本相较于1.0,进行了许多改进,主要是增加了多倍体物种的基因组调查,并提出Smudgeplot方法来估计基因组的倍性和基因组结构。
4.1. GenomeScope 2.0 使用步骤
- 上传第一步获得的k-mer频数分布表histo文件;
- 设置参数Kmer length为第一步选择的k-mer长度值,这里是17;
- 参数倍性Ploidy根据物种的倍性设定,默认是二倍体,设置成2;
- 参数Max k-mer coverage默认是-1,即不限制最大k-mer深度。建议按照物种情况修改,比如10000,以统计更准确。这个参数太小,可能造成过滤过多的Kmer,导致估计的基因组大小偏小的情况。这个参数太大则可能把高拷贝数量的DNA,比如叶绿体DNA,包括进Kmer的统计,造成GenomeScope算法的误差,所以还是不推荐使用-1或太大的值。
- 参数Average k-mer coverage for polyploid genome默认是-1,即不进行筛选,可以根据情况调整。
- 提交后几分钟就可以得到结果,保存结果图片可用于发表。
4.2. GenomeScope 2.0 结果
4.2.1. 二倍体结果
二倍体的GenomeScope 2.0 结果与GenomeScope 1.0 结果的主要不同之处在于杂合度结果(het)变成了2.0版本的代表基因型的aa和ab的比例,其中杂合基因型ab的比例即为杂合度。2.0结果中的p值代表设置的物种倍性。

Figure 2. GenomeScope2.0 二倍体结果示例
4.2.2. 区分异源四倍体和同源四倍体
GenomeScope2.0添加了参数倍性Ploidy,可以评估多倍体的基因组特征。
- 四倍体共有两种可能的拓扑结构,代表着同源四倍体和异源四倍体,每种拓扑包含三种杂合基因型和一种纯合基因型,共有五种基因型。(五倍体有五种可能的拓扑,六倍体有十六种)
- 根据结果中杂合基因型的分布模式可以区分异源四倍体和同源四倍体。
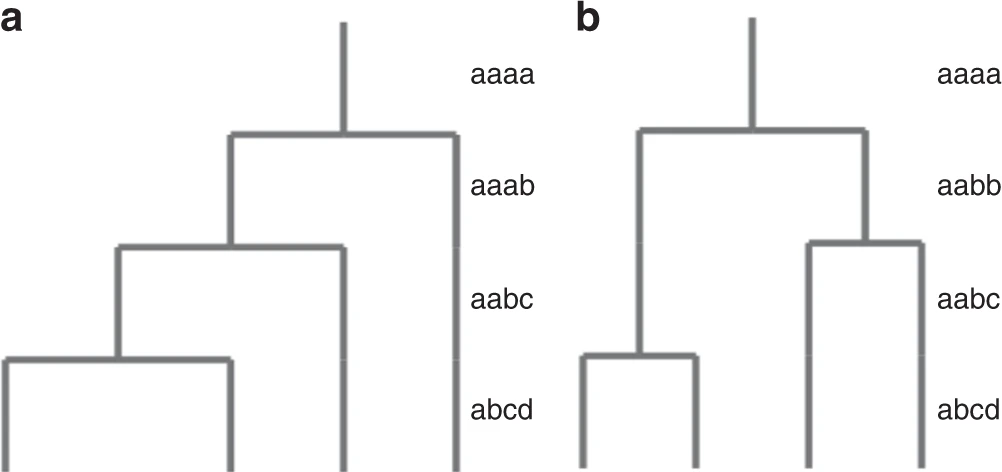
Figure 3. 四倍体的两种拓扑结构可能性 a异源四倍体,b同源四倍体
from GenomeScope 2.0 paper
- GenomeScope2.0的四倍体结果
在GenomeScope 2.0 的结果中,如果杂合基因型aaab的比例大于aabb,则认为该物种是异源四倍体;如果杂合基因型aaab的比例小于aabb,则认为该物种是同源四倍体。
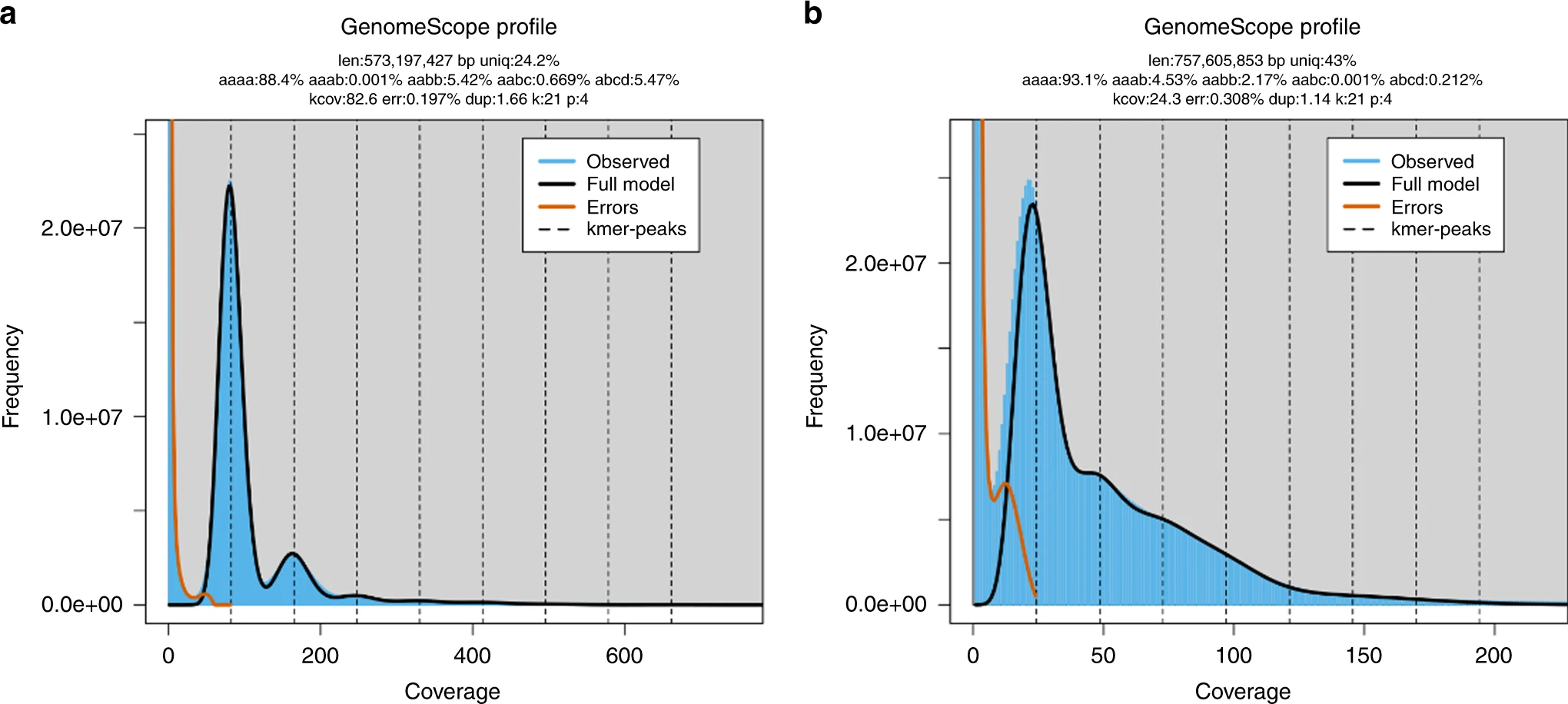
Figure 4. GenomeScope2.0 多倍体结果示例 a异源四倍体,b同源四倍体
from GenomeScope 2.0 paper
5. GenomeScope1.0本地版 —— 适用于二倍体物种
GenomeScope1.0的本地版是用一个R脚本实现的,在GenomeScope github可以下载genomescope.R脚本,下载后把genomescope.R文件加入环境变量即可使用。。
Rscript genomescope.R sample.histo k-mer_length read_length output_dir [kmer_max] [verbose]
- sample.histo:频数分布直方表,jellyfish的结果。
- k-mer_length:k-mer长度,通常是17,21,与jellyfish一致。
- read_length:reads长度,这里是150bp的PE reads,所以是150。
- output_dir:输出目录,结果图和文本都输出到这个目录。
6. GenomeScope2.0本地版 —— 适用于多倍体物种
6.1. 下载和安装
git clone https://github.com/tbenavi1/genomescope2.0.git #下载
cd genomescope2.0/
mkdir ~/R_libs #创建主目录下的R_libs文件夹用于安装本地R库
echo "R_LIBS=~/R_libs/" >> ~/.Renviron #创建/编辑.Renviron文件,使得R在创建的R_libs文件夹加载库
Rscript install.R #安装
安装后把目录下的genomescope.R文件加入环境变量即可使用。
6.2. 使用
genomescope.R -i histogram_file -o output_dir -k k-mer_length
- -i histogram_file:频数分布直方表,jellyfish或KMC的结果。
- -k k-mer_length:k-mer长度,通常是17,21,与jellyfish/KMC的设置一致。
- -o output_dir:输出目录,结果图和文本都输出到这个目录。
- -p ploidy:设置倍性。
- -l lambda:设置测序的平均k-mer覆盖率的初始猜测。
- -n name_prefix:设置输出文件的前缀。
- -m max_kmercov:设置从分析中排除高频k-mers的截止值,根据物种情况确定,推荐1000或10000。
7. GenomeScope实践经验
- 实际使用中发现,GenomeScope1.0和2.0常常估算差异较大。建议二倍体还是使用GenomeScope1.0。
- 在估算一个约300Mb的二倍体基因组时,GenomeScope1.0估算出来267Mb,GenomeScope2.0估算出来149Mb。
- 在估算一个约6Gb的四倍体基因组时发现,GenomeScope1.0估算出来5.5Gb,GenomeScope2.0估算出来2.7Gb。
8. Smudgeplot
Smudgeplot是2020年与GenomeScope2.0一起发表的用于估计物种的倍性的软件。开发者计划接下来把Smudgeplot整合进GenomeScope。
8.1. Smudgeplot原理
Smudgeplot从k-mer数据库中提取杂合k-mer对,然后训练杂合k-mer对。
通过比较k-mer对覆盖度的总数(CovA + CovB)和相对覆盖度(CovB / (CovA + CovB)),统计杂合k-mers对的数量,Smudgeplot可以解析基因组结构。
8.2. Smudgeplot安装
- 用于统计k-mers频数的软件。建议tbenavi1/KMC,里面包括一个smudge_pairs程序,用来找杂合k-mer对。也可以用jellyfish代替KMC,参考manual of smudgeplot with jellyfish。
- GenomeScope2.0
- 安装
conda install -c bioconda smudgeplot#conda安装
8.3. Smudgeplot使用
- 用KMC计算k-mer频率,生成k-mer直方图
mkdir tmp
ls *.fastq.gz > FILES
kmc -k21 -t16 -m64 -ci1 -cs10000 @FILES kmcdb tmp #计算k-mer频率
kmc_tools transform kmcdb histogram sample.histo -cx10000 #生成k-mer频数直方表sample.histo和k-mer直方图
kmc命令参数:
- -k21:k-mer长度设置为21
- -t16:线程16
- -m64:内存64G,设置使用RAM的大致数量,范围1-1024。
- -ci1 -cs10000:统计k-mer coverages覆盖度范围在[1-10000]的。
- @FILES:保存了输入文件列表的文件名为FILES
- kmcdb:KMC数据库的输出文件名前缀
- tmp:临时目录
kmc_tools命令参数:
- -cx10000:储存在直方图文件中counter的最大值。
- 选择覆盖阈值
- 可以目视检查k-mer直方图,选择覆盖阈值上(U)下(L)限。
- 也可以用命令估计覆盖阈值上(U)下(L)限。L的取值范围是[20-200],U的取值范围是[500-3000]。
L=$(smudgeplot.py cutoff kmcdb_k21.hist L)
U=$(smudgeplot.py cutoff kmcdb_k21.hist U)
echo $L $U # these need to be sane values
- 提取阈值范围的k-mers,计算k-mer pairs
用
kmc_tools提取k-mers,然后用KMC的smudge_pairs计算k-mer pairs。smudge_pairs比smudgeplot.py hetkmers使用更少内存,速度更快地寻找杂合k-mer pairs。kmc_tools transform kmcdb -ci"$L" -cx"$U" reduce kmcdb_L"$L"_U"$U"
smudge_pairs kmcdb_L"$L"_U"$U" kmcdb_L"$L"_U"$U"_coverages.tsv kmcdb_L"$L"_U"$U"_pairs.tsv > kmcdb_L"$L"_U"$U"_familysizes.tsv如果没有KMC,可以用
kmc_dump提取k-mers,然后用smudgeplot.py hetkmers计算k-mer pairs。kmc_tools transform kmcdb -ci"$L" -cx"$U" dump -s kmcdb_L"$L"_U"$U".dump
smudgeplot.py hetkmers -o kmcdb_L"$L"_U"$U" < kmcdb_L"$L"_U"$U".dump
- 生成污点图(smudgeplot)
smudgeplot.py plot kmcdb_L"$L"_U"$U"_coverages.tsv
生成两个基础的污点图,一个log尺度,一个线性尺度。
8.4. Smudgeplot结果
- 热度图,横坐标是相对覆盖度 (CovB / (CovA + CovB)) ,纵坐标是总覆盖度 (CovA + CovB) ,颜色是k-mer对的频率。
- 每个单倍型结构都在图上呈现一个”污点(smudge)”,污点的热度表示单倍型结构在基因组中出现的频率,频率最高的单倍型结构即为预测的物种倍性结果。(比如这个图提供了三倍体的证据,AAB的频率最高)

Figure 5. Smudgeplot污点图
from Smudgeplot github
8.5. Smudgeplot的KMC结果用于GenomeScope进行基因组调查
- 通过KMC获得的频数分布表结果可用于GenomeScope进行基因组调查
Rscript genomescope.R kmcdb_k21.hist <k-mer_length> <read_length> <output_dir> [kmer_max] [verbose]
9. references
Recommend
About Joyk
Aggregate valuable and interesting links.
Joyk means Joy of geeK