

用K-mer分析进行基因组调查(genome survey)
source link: https://yanzhongsino.github.io/2022/05/25/omics_genome.survey/
Go to the source link to view the article. You can view the picture content, updated content and better typesetting reading experience. If the link is broken, please click the button below to view the snapshot at that time.
1. 基因组调查(genome survey)
1.1. 基因组调查
基因组调查(genome survey)指基因组特征评估,一般指通过K-mer分析二代测序数据,获得基因组大小(genome size),杂合度(heterozygosity),重复序列比例,GC含量等基因组信息的手段。
基因组调查介绍
对于高等真核生物(特别是高等植物)来说,在进行基因组denovo测序和正式组装之前,首先构建DNA小片段文库进行中低深度的二代测序,使用测序所得的reads(通常是illumina的PE reads)进行基因组调查(genome survey),来初步评估基因组特征,包括基因组大小(genome size),杂合度(heterozygosity),重复序列比例,GC含量等,从而为后续的基因组测序、组装和结构注释方案提供参考依据。基因组调查的目的
基因组调查主要目的是获取两个方面的信息,一个是基因组的大小,一个是基因组复杂程度。
- 基因组大小(genome size)
因为测序费用是以测序量为单价计算,所以基因组越大,测序费用越高。 - 基因组复杂程度
基因组越复杂(杂合度越高,重复序列占比越高),意味着测序难度和组装难度越大。
- 基因组调查实践
- 为了准确估计基因组信息,建议测序深度为50X,即预估基因组大小的50倍,最小也不低于25X。
- 做基因组调查和后续做基因组denovo测序需要使用同一个个体,因为有些物种个体间的基因组特征有较大的差异,且同一个个体做survey的illumina数据可用于优化组装。
- 预估基因组大小可以通过已有研究粗略判断,包括流式细胞研究,近缘种研究,也推荐植物在C值数据库网站里查询。
- 基因组调查(genome survey)常常使用K-mer分析来实现。
1.2. 基因组复杂程度
基因组复杂程序的判断标准包括:基因组大小,倍性,杂合度,重复序列比例,GC含量等。
一般而言,基因组越大,重复序列比例越高; GC含量异常低或异常高,重复序列比例也会很高;多倍体基因组的杂合度高于二倍体。
判断基因组复杂程度可以参考以下经验性标准:
- 简单基因组: 单倍体;或纯合二倍体;或杂合度低于0.5%, 且重复序列低于50%, 且GC含量在35%-65%的二倍体。
- 复杂基因组: 杂合度在0.5%~1.2%之间,或重复序列高于50%,或GC含量异常(<35%或>65%)的二倍体,或者多倍体。
- 高复杂基因组: 杂合度>1.2%;或重复序列占比大于65%。
2. K-mer分析
K-mer分析可以用在生物信息学许多方面,这篇博客的K-mer分析特指用于基因组调查的K-mer分析方法。
2.1. K-mer相关概念
- monomeric unit (mer): 单体单元,单位是nt或者bp。通常用于双链核酸中的单位,100 mer DNA相当于每一条链有100nt,那么整条链就是100bp。
- K-mer概念
在生物信息学中,K-mer是指包含在一段序列中的长度为k的子串。一段长度为L的核酸序列,以一个碱基为步长滑动,一共可以生成(L-K+1)个K-mers,另外还可以用这段核酸的反向互补序列再生成一次K-mer。
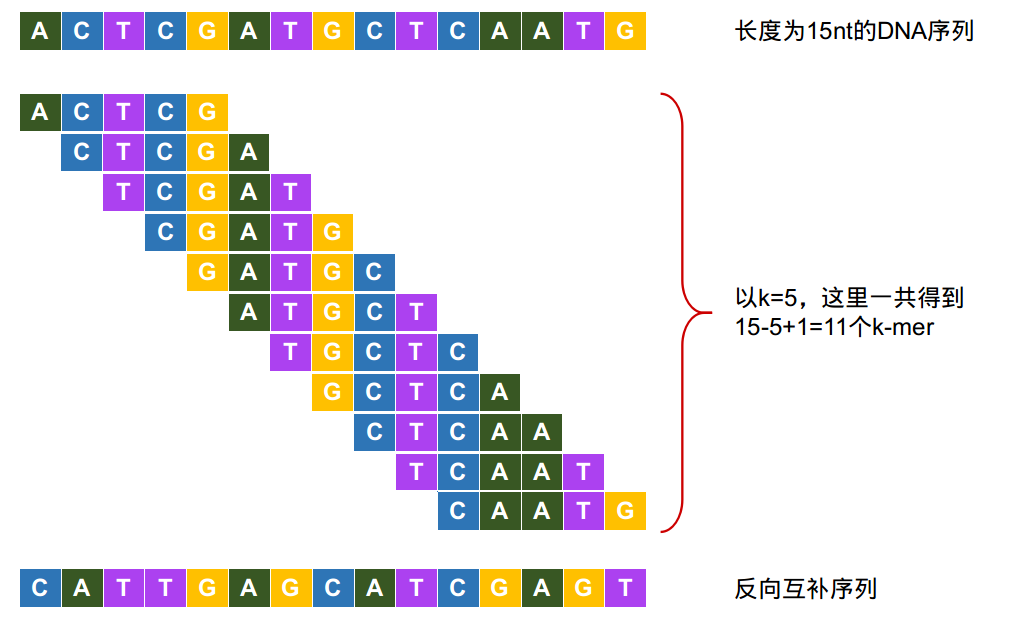
Figure 1. K-mer示例
from 博客:k-mer与基因组组装
2.2. K-mer分析步骤
- 通过切割二代测序的reads为K-mers
- 统计K-mer的总数和每一种K-mer的频数
- 绘制K-mer的频数分布图
- 根据K-mer的频数分布的主峰峰值判定K-mer的期望深度(即主峰对应的K-mer频数)。
- 根据K-mer的期望深度和K-mer的总数估计基因组大小。
- 根据低频K-mer估计数据错误率,并修正基因组大小的估计。
- 根据K-mer的其他峰估计K-mer的杂合度和重复序列比例。
2.3. K-mer原理
K-mer分析应用的前提假设是测序的reads是随机分布在基因组上的。
首先定义几个变量,方便解释原理:
- 基因组大小:G
- read读长:L
- 总reads条数:n
- K-mer长度:K
2.3.1. 碱基深度分布
- 单条read测序覆盖到某一个碱基的概率:L/G。
- 因为L/G很小,n很大,每个碱基覆盖深度服从泊松分布。
- 则每个碱基的覆盖深度的期望为:d=(L/G)*n。
2.3.2. K-mer深度分布
- 一个大小为G的基因组可以产生的K-mer种类约为G
- 假设一个基因组产生的K都是unique的,从一个大小为G的基因组可以得到(G-K+1)种不同的K-mer,
- 一般而言,基因组大小G在几百Mb或者Gb为单位,远大于K和1,所以K和1可以忽略不计,约等于G种K-mer。
- 单条read测序完全覆盖某种K-mer的概率:(L-K+1)/G
- K-mer种类的总数约为G
- 单条read能产生的K-mer种类数量为(L-K+1)
- 假设read随机分布在基因组上,单条read测序完全覆盖某种K-mer的概率就为:(L-K+1)/G
- 同样因为(L-K+1)/G很小,n很大,每种K-mer的覆盖深度服从泊松分布。
- 每种K-mer的覆盖深度的期望为:D=((L-K+1)/G)*n
- 由此可以得到,基因组大小为:G=(L-K+1)*n/D
2.3.3. 基因组大小
- 计算总K-mer个数N和K-mer期望深度D
- 对测序reads进行K-mer分割,获得总K-mer个数N。
- 统计所有分割的K-mer,绘制频数分布图。
- 理想情况下(不考虑测序错误、序列重复性和杂合序列的条件下),K-mer的频数分布遵循泊松分布,可以将频数分布峰值的K-mer频数作为K-mer的期望深度D。
- 通过下面公式可知,基因组大小G=N/D。
只要通过K-mer分析计算得到K-mer的总数量N和K-mer的期望深度D,则可以算出基因组大小G。
- K-mer的总数量N=(L-K+1)*n
- K-mer的期望深度D=((L-K+1)/G)*n
2.3.4. 基因组调查
在不考虑测序错误、序列重复性和杂合序列的条件下,K-mer的深度分布遵循泊松分布。但实际情况是三者都存在,所以需要计算错误率,重复序列占比和杂合度,并根据计算结果修正对基因组大小的估计。
- 测序错误:一般认为低频K-mer(K=1,2…)是测序错误引起的,去除低频K-mer并计算错误率以修正基因组大小的估计。
- 一般把拐点前的低频K-mer当作错误去除。
- 重复序列占比
- 基因组中存在的重复序列会使对应的K-mer频数高于K-mer期望深度D。
- 在K-mer频数分布图中重复序列对应的K-mer会表现为频数大于主峰的一个重复峰。
- 根据主峰右侧的重复峰可以估计基因组的重复序列比例。
- 通过模型计算一个阈值,比如阈值为1.6倍主峰,理论上单拷贝序列(非重复序列)出现在1.6倍主峰右边的概率很低,可以把1.6倍主峰之后的看作重复K-mer,统计重复K-mer的总数量m,从而可以算出:重复序列的大小R=重复K-mer的总数量m/K-mer的期望深度D。
- R/G即为重复序列占基因组的比例。
- 基因组中存在的杂合序列会对应两种类型的K-mer,并使得这些K-mer频数低于K-mer期望深度D。
- 假设两种K-mer数量大致相等,在K-mer频数分布图中杂合序列对应的K-mer会表现为频数约为主峰一半的一个杂合峰。
- 根据主峰左侧的杂合峰可以估计基因组的杂合度。
假设基因组中的杂合率为 h(每个碱基为杂合点的概率),一个K-mer是纯合k-mer的概率为 P1 = (1-h)^k, 则是杂合的概率 P2 = 1-P1 = 1-(1-h)^k。
假设产生的K-mer种类的总数为M,其中属于单拷贝序列(非重复序列)的K-mer种类
在二倍体中,单拷贝序列(非重复序列)的同源区域,会产生 U×P2 的杂合K-mer,这个数是可以通过计算得到的:只要统计非重复K-mer的总数 M , M-U即为产生的杂合K-mer数。则 M-U = U×P2 = U×(1-(1-h)^k)。
通过计算,即可通过M和U的值得到基因组的杂合率h。
在实际应用过程中,估计了基因组的错误率、杂合度和重复序列比例后,重新修正基因组大小的估计,从而得到基因组调查的结果。

Figure 2. K-mer分析(GenomeScope)结果示例
2.4. K-mer用途
许多分析都会用到K-mer的处理方法,把测序得到的reads通过截取K-mer后用于分析。比如评估基因组特征,组装基因组,物种样品污染评估等。评估基因组特征(genome survey)包括评估基因组大小(size),杂合度,重复序列比例等。
组装基因组
组装基因组使用K-mer的目的主要是去除低频率的K-mer以提高组装结果准确性。评估基因组大小(size)
在K-mer原理部分解释了用K-mer评估基因组大小的公式:
基因组大小G=总K-mer数量N/K-mer期望深度D
在不考虑测序错误、序列重复性的条件下,K-mer的深度分布遵循泊松分布,可以将深度分布曲线的峰值作为期望测序深度。
评估基因组杂合度
根据K-mer频数分布图的杂合峰,可以估计基因组的杂合度。评估基因组重复序列比例
根据K-mer频数分布图的重复峰,可以估计基因组的重复序列比例。评估物种样品污染
根据K-mer频数分布图,可以评估测序样品污染情况。
- 如果K-mer频数分布图出现两个明显的峰,但两个峰的横坐标又不是二倍关系,那就可能是DNA污染导致的。
- 因为一个物种的杂合峰的频数约为主峰的一半,重复峰的频数约为主峰的两倍。
- 如果DNA中存在两个物种的样品(即有污染),期望会出现对应的两个峰,且两个峰的横坐标之间也非两倍的关系,由此可以初步判定该测序样本存在污染。
- 至于具体是因为什么物种造成污染,则可以通过blast比对nr库进行简单的判断。
除了通过K-mer频数分布评估DNA样品的污染程度外,还可以通过GC含量分布图判断,查看图中是否存在多个密度集中的类群。
2.5. K-mer的特点
- 增加准确率
- 二代测序的准确率已达到99.9%,但测序量非常大时,错误碱基的绝对数量(比如10亿碱基里错误碱基数量会达到1000万个)还是会对分析有很大的影响。
- 由于测序错误具有随机性,通过将reads切割产生的K-mer中,测序错误生成的K-mer绝大多数都是测序物种中不存在的K-mer,因此都只出现1次(或很少的几次),要是将这些低频的K-mer去掉,就有较大可能去除测序错误,从而使得分析(基因组调查,组装基因组)结果更可靠。
- 不适用过于复杂的基因组
- K-mer分析适用于分析唯一主峰区域所占比例较大的基因组,当基因组杂合非常高或者重复序列比例非常大时,其影响可能导致无法通过K-mer分析正确估计基因组大小。
2.6. K-mer的选择
K-mer的大小选择:K应该足够大到K-mer可以映射到基因组的唯一位置;但太大的K-mer会降低去除低频K-mer代表的错误碱基的概率(增加错误率),也会降低K-mer深度(使得K-mer频数分布的峰不明显),而且大的K-mer会增加计算资源的使用。基因组调查一般选17,21比较常见。
- K-mer只能是奇数?
把K-mer设置成奇数是为了防止通过K-mer组装时,正反链混淆。偶数的K-mer的反向互补序列常常与自身一样,从而组装K-mer时会混淆正反链的组装,奇数的K-mer就不存在这个问题。 - K-mer的长度代表了可能存在的K-mer种类的数量(4的K次方),越长的K-mer片段映射的物种特异性越强。基因组越大,需要的K-mer越长。
- 当基因组中有较多重复序列时,可以用较大的K-mer来跨过高重复的区域,从而获得更加准确及完整的基因组草图;
- 由于reads上的碱基错误率的存在,选择较长的K-mer会带来较高的错误率,这也可以加大测序深度来弥补。
- 如果是用于组装基因组,为了得到更加完整的基因组,要尽可能使用较长的K-mer用于组装。
3. K-mer分析软件概况
K-mer分析分为K-mer频数统计和基因组特征评估两步。
- jellyfish
jellyfish可以实现第一步K-mer频数统计。特点是使用Hash表存储数据,能多线程运行;速度快,内存消耗小。 - GenomeScope
软件GenomeScope可以利用K-mer频数统计结果进行基因组特征评估。 - KAT(The K-mer Analysis Toolkit)
软件KAT(The K-mer Analysis Toolkit)可以实现两步。包含多个工具来帮助用户通过使用k-mer对测序数据进行简单分析,如组装完整性、测序错误、是否有污染等。 - gce
可以分别实现两步骤。 - KmerGenie
软件KmerGenie可以同时实现两步。最大优点在于可以实现在多个预设K-mer下的自动分析,除了进行常规的k-mer频数统计之外,还能够基于不同k-mer自动计算基因组大小,并为基因组组装评估一个最佳组装k-mer数值作为备选。
notes:
- 推荐使用jellyfish+GenomeScope进行K-mer分析。
- K-mer长度常用17/21。
- 软件KmerGenie,gce和jellyfish获取的频数分布表,都可用于软件genomescope和gce第二步骤的分析。
- 由于gce第一步骤支持的最大K-mer频数为255,大于255的数据被合并;而jellyfish统计到10000行,预估结果会更为准确。
- GenomeScope对于高重复序列的基因组统计的基因组大小会偏小,建议max kmer coverage设置大一点,大于等于10000。
- 有些软件有另一个参数需注意和设定,单倍体模式还是杂合模式,可以两种模式都分析,查看差别。
- 实践经验发现,K-mer值设置得越高,估计出来的基因组size会越大;
- 另外,在jellyfish里的jellyfish histo统计频数分布时,用参数-h 10000把统计上限调高,以及在GenomeScope阶段,Max kmer coverage设置的大一些(即统计进的kmer数量越多),估算出来的基因组大小也会略大一些。
4. references
Recommend
About Joyk
Aggregate valuable and interesting links.
Joyk means Joy of geeK