

使用 ggtree 实现进化树的可视化和注释
source link: https://cosx.org/2015/11/to-achieve-the-visualization-and-annotation-of-evolutionary-tree-using-ggtree/
Go to the source link to view the article. You can view the picture content, updated content and better typesetting reading experience. If the link is broken, please click the button below to view the snapshot at that time.
本文作者:余光创,目前就读于香港大学公共卫生系,开发过多个 R/Bioconductor 包,包括 ChIPseeker, clusterProfiler, DOSE, ggtree, GOSemSim 和 ReactomePA。
进化树看起来和层次聚类很像。有必要解释一下两者的一些区别。
层次聚类的侧重点在于分类,把距离近的聚在一起。而进化树的构建可以说也是一个聚类过程,但侧重点在于推测进化关系和进化距离 (evolutionary distance)。
层次聚类的输入是距离,比如 euclidean 或 manhattan 距离。把距离近的聚在一起。而进化树推断是从生物序列(DNA 或氨基酸)的比对开始。最简单的方法是计算一下序列中不匹配的数目,称之为 hamming distance(通常用序列长度做归一化),使用距离当然也可以应用层次聚类的方法。进化树的构建最简单的方法是非加权配对平均法(Unweighted Pair Group Method with Arithmetic Mean, UPGMA),这其实是使用 average linkage 的层次聚类。这种方法在进化树推断上现在基本没人用。更为常用的是邻接法(neighbor joining),两个节点距离其它节点都比较远,而这两个节点又比较近,它们就是 neighbor,可以看出 neighbor 不一定是距离最近的两个节点。真正做进化的人,这个方法也基本不用。现在主流的方法是最大似然法 (Maximum likelihood, ML),通过进化模型(evolutionary model) 估计拓朴结构和分支长度,估计的结果具有最高的概率能够产生观测数据(多序列比对)。另外还有最大简约法和贝叶斯推断等方法用于构建进化树。
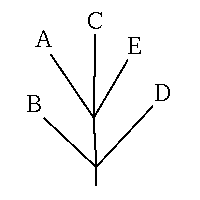
是最常用的存储进化树的文件格式,如上面这个树,拓朴结构用 newick 格式可以表示为:
(B,(A,C,E),D);
括号最外层是根节点,它有三个子节点,B, (A,C,E)和 D,而节点 (A,C,E) 也有三个子节点 A,C 和 E。
加上分支长度,使用: 来分隔:
(B:6.0,(A:5.0,C:3.0,E:4.0):5.0,D:11.0);
比如 A:5.0 代表的是 A 与其父节点的距离是 5.0。
内部节点也可以有 label,写在相应的括号外面,如下所示:
(B:6.0,(A:5.0,C:3.0,E:4.0)Ancestor1:5.0,D:11.0);
这是最为广泛支持的文件格式,很多进化树可视软件只支持 newick 格式。
ggtree 的开发源自于我需要在树上做注释,发现并没有软件可以很容易地实现,通常情况下我们把统计信息加到节点的 label 上来展示,比如 CodeML 的 dN/dS 分析,输出文件里就给用户准备了 newick 树文本,把 dN/dS ( ω ) 加于节点 label 之上:
codeml_file <-system.file("extdata/PAML_Codeml/mlc", package="ggtree")
tree_text <-readLines(codeml_file)[375:376]
tree_text
# [1] "w ratios as labels for TreeView:"
# [2] "(K #0.0224 , N #0.0095 , (D #0.0385 , (L #0.0001 , (J #0.0457 , (G #0.1621 , ((C #0.0461 , (E #0.0641 , O #0.0538 ) #0.0001 ) #0.0395 , (H #0.1028 , (I #0.0001 , (B #0.0001 , (A #0.0646 , (F #0.2980 , M #0.0738 ) #0.0453 ) #0.0863 ) #1.5591 ) #0.0001 ) #0.0001 ) #0.0549 ) #0.0419 ) #0.0001 ) #0.0964 ) #0.0129 );"
这种做法只能展示一元信息,而且修改节点 label 真心是个脏活,满满的都是不爽,我心中理想的方式是树与注释信息分开,注释信息可以方便地通过图层加上去,而且可以自由组合。于是着手开发 ggtree 是个简单易用的 R 包,一行代码
ggtree(read.tree(file))
即可实现树的可视化。而注释通过图层来实现,多个图层可以完成复杂的注释,这得力于 ggtree 的设计。其中最重要的一点是如何来解析进化树。
ggtree 的设计
进化树的解析
除了 ggtree 之外,我所了解到的其它画树软件在画树的时候都把树当成是线条的集合。很明显画出来的进化树就是在画一堆线条,但是线条表示的是父节点和子节点的关系,除此之外没有任何意义,而节点在进化树上代表物种,叶子节点是我们构建进化树的物种,内部节点是根据叶子节点推断的共同祖先。我们所有的进化分析、推断、实验都是针对节点,节点才是进化树上有意义的实体。这是 ggtree 设计的基础,ggtree 只映射节点到坐标系统中,而线条在 geom_tree 图层中计算并画出来。这是与其它软件最根本的不同,也是 ggtree 能够简单地用图层加注释信息的基础。
扩展 ggplot2
有很多可视化包基于 ggplot2 实现,包括各种 gg 打头的,号称扩展了 ggplot2,支持图形语法 (grammar of graphics),我并不认同。虽然基于 ggplot2 产生的图,我们可以用 theme 来进一步调整细节,用 scale_系列函数来调整颜色和标尺的映射,但这些不足以称之为支持图形语法,图形语法最关键核心的部分我认为是图层和映射。
像 ggphylo, OutbreakTools 和 phyloseq 这几个包都有基于 ggplot2 的画树函数,但其实都不支持图形语法,它们所实现的是复杂的函数,画完就完事了,用户并不能使用图层来添加相关的信息。
如果在 OutbreakTools 这个包中:
if (show.tip.label) {
p <- p + geom_text(data = df.tip, aes(x = x, y = y, label = label),
hjust = 0, size = tip.label.size)
}
如果 show.tip.label=FALSE,当函数返回 p 时 df.tip 就被扔掉,用户想要再加 tip.label 就不可能了。 ggphylo 和 phyloseq 都是类似的实现,这些包把树解析为线条,所以节点相关的信息需要额外的 data.frame 来存储,并且只有极少数的预设参数,比如上面例子中的 tip.label。在上面的例子中,用户连更改 tip.label 的颜色都不可能,更别说使用额外的注释信息了。
这几个包所实现的画图函数,都可以很容易地用 ggtree 实现,并用经过测试,ggtree 运行速度比这几个包都要快。更多信息请参考 ggtree 的 wiki 页面。
ggtree 是真正扩展 ggplot2,支持图形语法的包。我们首先扩展 ggplot 支持 tree object 做为输入,并实现 geom_tree 图层来画线条。
library(ggplot2)
library(ggtree)
set.seed(2015-11-26)
tree <-rtree(30)
ggplot(tree, aes(x, y)) + geom_tree()
ggtree 函数是 ggplot() + geom_tree() + xlab(NA) + ylab(NA) + theme_tree() 的简单组合。
ggtree(tree)
想要加 tip.label,用 geom_tiplab 图层,并且 ggplot2 的图层都可以直接应用 ggtree。
ggtree(tree) + geom_tiplab() + geom_point(color='firebrick')
 树的操作与注释
树的操作与注释
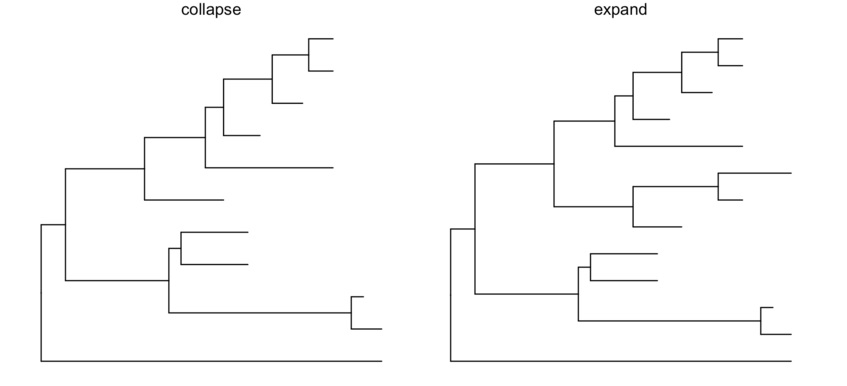
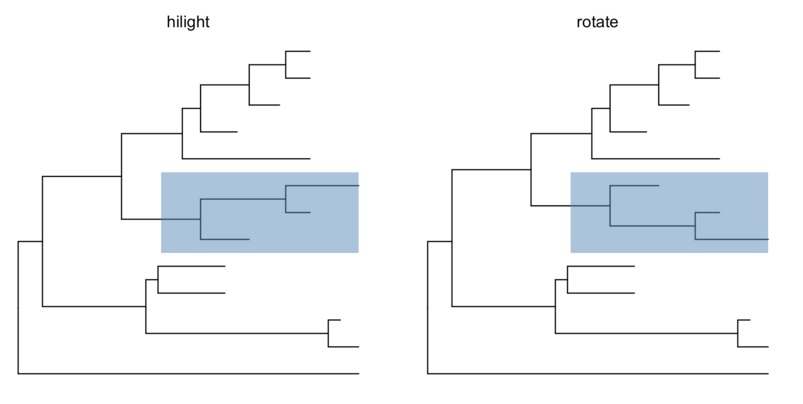
ggtree 提供了多个函数可以把 clade 放大缩小 (scaleClade),折叠(collapse) 和展开(expand),位置调换和旋转,以及分类(groupOTU, groupClade)。
nwk <- system.file("extdata", "sample.nwk", package="ggtree")
tree <- read.tree(nwk)
p <- ggtree(tree)
cp <- ggtree(tree) %>% collapse(node=21) + ggtitle('collapse')
ep <- cp + expand(node=21) + ggtitle('expand')
hp <- p + hilight(node=21) + ggtitle('hilight')
rp <- hp + rotate(node=21) + ggtitle('rotate')
library(gridExtra)
grid.arrange(cp, ep, hp, rp, ncol=2)
支持多种文件格式
ggtree 支持的文件格式包括 Newick, Nexus, NHX 和 jplace。
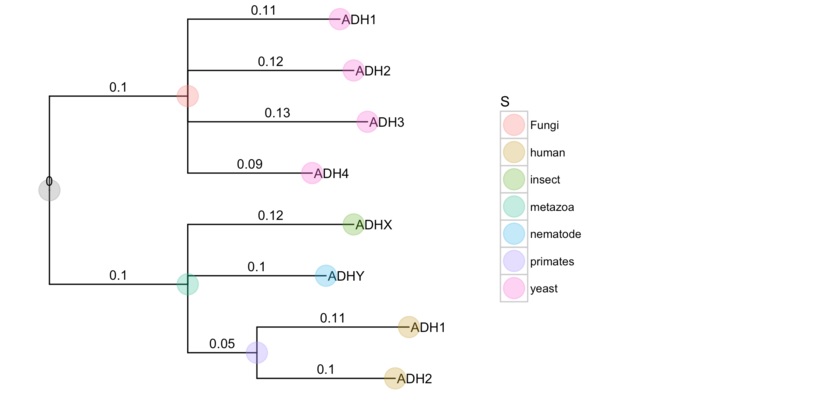
上面已经展示了 Newick 格式,下面的例子是 NHX 格式:
nhxfile = system.file("extdata", "ADH.nhx", package="ggtree")
nhx <-read.nhx(nhxfile)
ggtree(nhx, ladderize=F) + geom_tiplab() + geom_point(aes(color=S), size=8, alpha=.3) +
theme(legend.position="right") +
geom_text(aes(label=branch.length, x=branch), vjust=-.5) +
xlim(NA, 0.3)
 支持解析多种软件的输出文件
支持解析多种软件的输出文件
我们知道 FigTree 是针对 BEAST 的输出设计的,可以把 BEAST 的统计推断拿来给树做注释,但很多的进化分析软件并没有相应的画树软件支持,用户很难把信息展示出来。
ggtree 支持 ape, phangorn, r8s, RAxML, PAML, HYPHY, EPA, pplacer 和 BEAST 的输出。相应的统计分析结果可以应用于树的注释。可以说 ggtree 把这些软件分析的结果带给了 R 用户,通过 ggtree 的解析, 这些进化分析结果可以进一点在 R 里进行处理和统计分析,并不单单是在 ggtree 中展示而已。
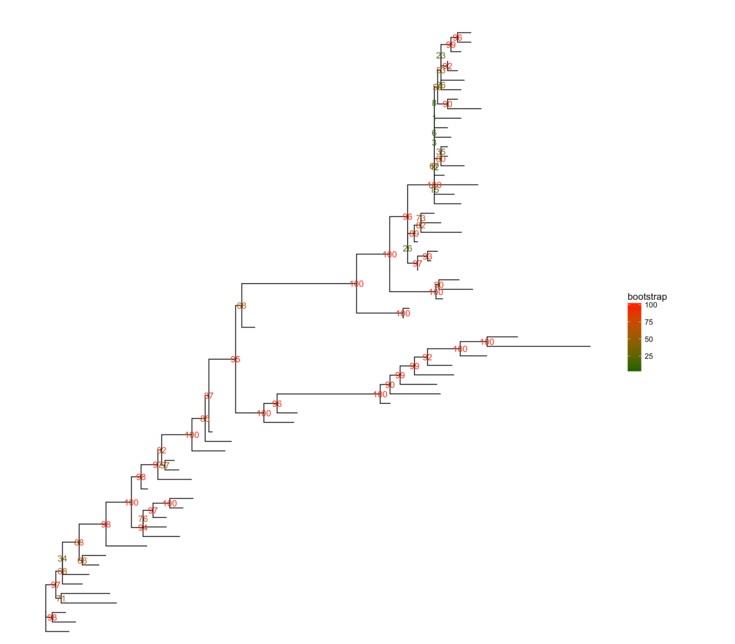
RAxML bootstrap 分析
raxml_file <-system.file("extdata/RAxML", "RAxML_bipartitionsBranchLabels.H3", package="ggtree")
raxml <-read.raxml(raxml_file)
ggtree(raxml) + geom_text(aes(label=bootstrap, color=bootstrap)) +
scale_color_gradient(high='red', low='darkgreen') +
theme(legend.position='right')
multiPhylo 也是支持的,所以 100 颗 bootstrap 树可以同时用一行代码展示出来。
btree_file <-system.file("extdata/RAxML", "RAxML_bootstrap.H3", package="ggtree")
btree = read.tree(btree_file)
ggtree(btree) + facet_wrap(~.id, ncol=10)

如果不分面,这 100 颗树会重叠画在一起,这也能很好地展示 bootstrap 分析的结果,bootstrap 值低的 clade,线条会比较乱,而 bootstrap 值高的地方,线条一致性比较好。
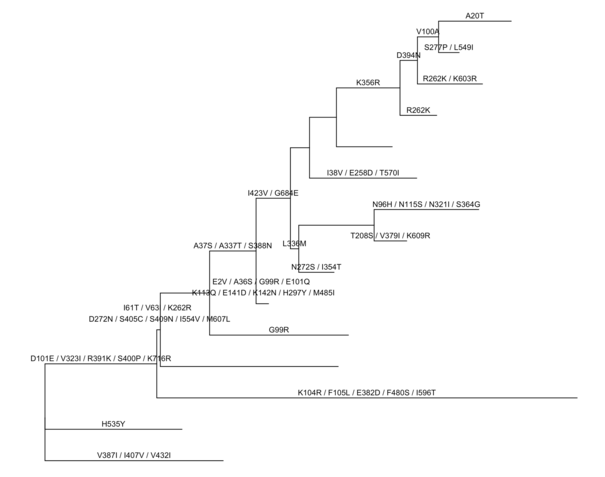
使用 BaseML 预测的祖先序列,ggtree 解析结果的同时会把父节点到子节点的 subsitution 给统计出来,可以直接在树上注释:
rstfile <-system.file("extdata/PAML_Baseml", "rst", package="ggtree")
rst <-read.paml_rst(rstfile)
p <-ggtree(rst) + geom_text(aes(label=marginal_AA_subs, x=branch), vjust=-.5)
print(p)
不同于 BaseML 以碱基为单位,CodeML 预测祖先序列,以密码子为单位。g>ree 定义了一个操作符 %<%,如果有相同的注释信息要展示,可以用 tree object 来更新 tree view。
rstfile <-system.file("extdata/PAML_Codeml", "rst", package="ggtree")
crst <-read.paml_rst(rstfile)
p %<% crst
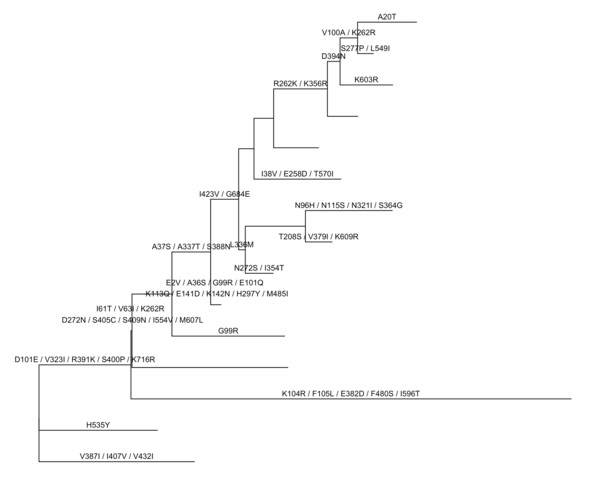
像上面的例子,用 crst 来更新 p,就是用 crst 画出来的树 + 注释。对比两图,可以发现 BaseML 和 CodeML 推测的祖先序列是稍有不同的。
CodeML 的 dN/dS 分析,我们可以直接把数据拿来给树上色。同样道理分类数据也可以拿来上色。
mlc_file <-system.file("examples/mlc", package="ggtree")
mlc <-read.codeml_mlc(mlc_file)
ggtree(mlc, aes(color=dN_vs_dS)) +
scale_color_continuous(limits=c(0, 1.5), high='red', low='green', oob=scales::squish, name='dN/dS') +
theme(legend.position='right')
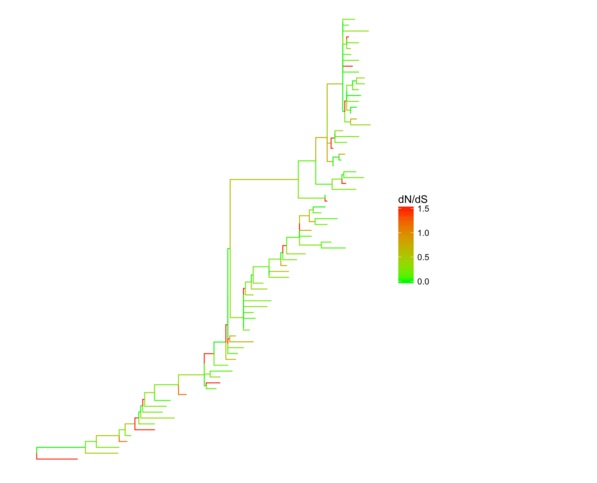 使用用户定义数据
使用用户定义数据
进化树已经被广泛应用于各种跨学科的研究中,随着实验技术的发展,各种数据也更易于获得,使用用户数据注释进化树,也是 ggtree 所支持的。
nwk <-system.file("extdata", "sample.nwk", package="ggtree")
tree <-read.tree(nwk)
p <-ggtree(tree)
dd <-data.frame(taxa = LETTERS[1:13],
place = c(rep("GZ", 5), rep("HK", 3), rep("CZ", 4), NA),
value = round(abs(rnorm(13, mean=70, sd=10)), digits=1))
## you don't need to order the data
## data was reshuffled just for demonstration
dd <-dd[sample(1:13, 13), ]
row.names(dd) <- NULL
print(dd)

在上面的例子中,使用一个分类数据和一个连续型数据,输入的唯一要求是第一列是 taxon label。ggtree 中定义了操作符 %<+%,来添加数据。添加之后,用户的数据对 ggplot 是可见的。可以用于树的注释。
p <- p %<+% dd + geom_text(aes(color=place, label=label), hjust=-0.5) +
geom_tippoint(aes(size=value, shape=place, color=place), alpha=0.25)
p+theme(legend.position="right")
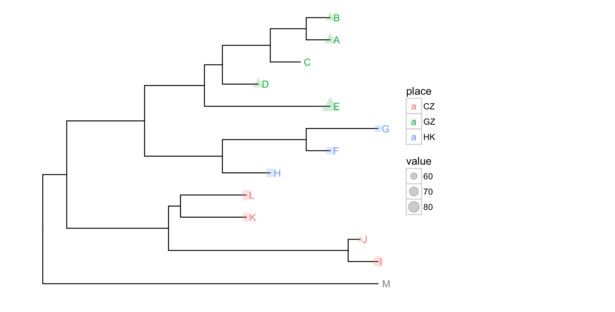
ggtree 还支持用户把自己的数据和树保存为 jplace 格式。
更多的实例请参考 vignette。
ggtree 允许把不同软件的分析结果整合在一起,同时在树上展示或者比较结果。在我们提交的论文中,使用了整合 BEAST 和 CodeML 的例子,在树上展示 dN/dS、时间轴、氨基酸替换、clade support values、物种和基因型 (genotype)等多维信息,6 种不同的信息同时展示在一颗进化树上,这是个复杂的例子,我们在附件 1 中展示了可重复的代码。如果有兴趣,可以留意一下我们的文章。 🙂
其它好玩的功能
我们把树当成节点的集合,而不是线条的集合,这一点回归到了进化树的本质意义上,使这一实现成为可能。而支持图形语法,与 ggplot2 的无缝衔接又让注释变得更加容易 ggtree 为我们打开了各种注释和操作的可能性。甚至于可以创造出好玩的图,比如使用 showtext 来加载图形化的字体、 用 emoji 来画树、使用 图片来注释树等等。
一个比较正经又好玩的是使用 PhyloPic 数据库上的图形。
pp <-ggtree(tree) %<+% phylopic("79ad5f09-cf21-4c89-8e7d-0c82a00ce728", color="steelblue", alpha = .3)
pp + geom_tiplab(align=T, linetype='dashed', linesize=.5) + geom_tippoint(color='firebrick', size=2)
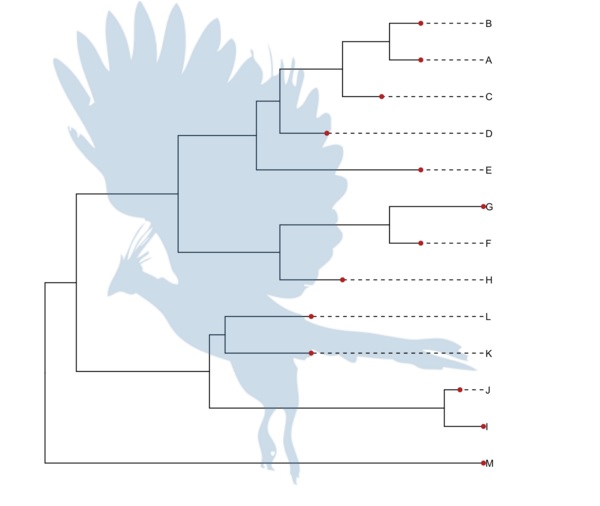
另一个好玩又为我们展现各种可能性的是 subview 函数,它使得图上加小图变得特别容易。并且已经被应用于地图上加饼图。解决这个问题的初衷在于,想要给节点加饼图注释。有了 subview 函数之后,这会变得很容易,当然我还没有写出给节点加饼图的函数,因为我还没有这个需求,得有一些实际的数据做参考,这样才能够设计出更易用的函数呈现给用户。
很多的功能还在开发之中,有问题 / 建议请及时在 Github 上报告 (中英文都可以)。
余光创,香港大学公共卫生学院,生物信息学博士生。博客:https://guangchuangyu.github.io, 公众号:biobabble
敬告各位友媒,如需转载,请与统计之都小编联系(直接留言或发至邮箱:[email protected]),获准转载的请在显著位置注明作者和出处(转载自:统计之都),并在文章结尾处附上统计之都微信二维码。

Recommend
About Joyk
Aggregate valuable and interesting links.
Joyk means Joy of geeK