

新冠病毒将如何变异?机器学习给你答案
source link: http://ai.51cto.com/art/202004/614736.htm
Go to the source link to view the article. You can view the picture content, updated content and better typesetting reading experience. If the link is broken, please click the button below to view the snapshot at that time.
本文转载自公众号“读芯术”(ID:AI_Discovery)
病毒和其他微生物一样,为了在地球上存活,不断进化和变异。这对于人类来说就有点儿可怕了,尤其是新冠病毒肆虐全球的今天,它还想整什么幺蛾子?

事实上,变异已经发生了。先来看一段由人类病毒进化而来的蝙蝠病毒的RNA核苷酸序列:
AAAATCAAAGCTTGTGTTGAAGAAGTTACAACAACTCTGGAAGAAACTAAGTT
以及一段新冠病毒的RNA核苷酸序列:
AAAATTAAGGCTTGCATTGATGAGGTTACCACAACACTGGAAGAAACTAAGTT
显然,新冠病毒为了适应新宿主,其原始结构已发生改变。准确来说,已经有20%的原始结构发生变异,但因大部分结构并未改变,所以病毒还未发生变种。
研究者发现新冠病毒已发生重复变异以持续存活。在与新冠病毒的较量中,我们不仅要知道如何消灭病毒,还要了解病毒如何变异以及怎样应对病毒变异。本文将尝试用K-Means和PCA探究这一点。
什么是基因组序列?
如果您对RNA核苷酸序列有所了解,那么可直接略过这部分内容。
基因组序列,通常我们称之为“解码”,是对样本进行DNA分析的重要步骤。一般来说,正常细胞中有23对携带DNA结构的染色体。

DNA为双螺旋结构,解开后呈梯形,构成梯形的是成对出现的碱基。DNA有四种碱基,分别是:腺嘌呤、胸腺嘧啶、鸟嘌呤和胞嘧啶。其中,腺嘌呤只和胸腺嘧啶配对,鸟嘌呤只和胞嘧啶配对。这四种碱基分别用A、T、G、C表示。
这些碱基对通过排列组合可以决定生物体蛋白质的具体结构,也就是从本质上决定病毒如何作用的DNA。

通过使用特殊仪器,比如排序仪器和一些特殊的标记法,可以揭开某特殊片段DNA序列的神秘面纱。而由此获得的信息可以进一步地分析和比较,有助于帮助研究者识别基因变化、疾病和表型相关以及判断药物靶标。
基因组序列,由A、T、G和C组成的长链,是生物体对自然环境的具体表现。生物体的变异是通过改变DNA来完成的。研究基因组序列是分析病毒变异的有效方式。
了解数据
以下数据可在 Kaggle找到:

每一行数据表示蝙蝠病毒发生的一次变异。仅仅几周时间,新冠病毒为提高存活率已经发生了262次变异。
一些重要数据:
- query acc.ver表示原始病毒的标示符。
- Mismatches表示变异病毒和原始病毒的不同项的数量
- subject acc.ver 表示变异病毒的标示符。
- % identity表示原始病毒和变异病毒的相似程度。
- alignment length 表示序列中相同或形近数目的具体数量。
- bit score表示形近度,分数越高,形近度越高。
下图为一些数据每列的统计数值(此数据用Python通过data.describe()便可轻松得出):
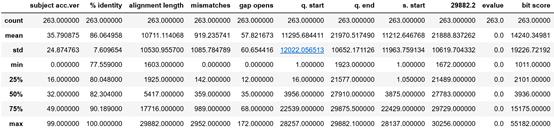
通过观察 % identity 列的数据,我们可以发现一个有趣的现象,每次变异的最小比对值大约为77.6%。对于这列数据而言,7%的标准偏差已经算相当大了,而这么大的标准差也就意味着变异范围的扩大。bit score的值也表明标准偏差已经很大了,竟然比均值还大!
关联热图是将数据可视化的好方法。每一单元格都表明了各个特性之间的关联。

许多数据彼此间高度关联,因为大多数数值的变化是相互影响的。这里需要特别注意的是alignment length 和 bit score的高度关联性。
运用K-Means创造变异集群
K-Means是一种应用于机器学习的聚类算法,可发现未来空间内的数据点群。K-Means的目标是发现变异集群,以此来为病毒本质的研究和处理提供依据。
然而,我们仍然需要选择集群k的数量。虽然这和在二维中绘制点一样简单,但在高维中是无法实现的(如果我们想保留大部分信息的话)。运用肘部方法选择k过于主观、不准确,所以我们将使用剪影法。

剪影法是k个聚类的分数,说明聚类对数据的适应程度。Python中的sklearn库令使用K-Means和silouhette方法变得非常简单。
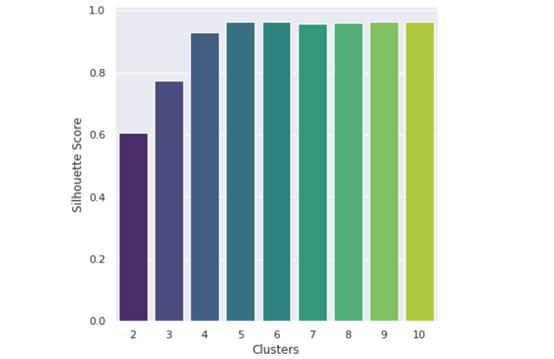
似乎5个集群对数据来说是最好的。现在,我们可以确定集群中心了。这些是每个集群围绕的点,代表了(在本例中)5种主要突变类型的数值评估。

Note: The features have been standardized to put them all on the samescale. Otherwise, columns would not be comparable.
此热图中的每列表示每个群集的属性。因为这些点是按比例缩放的,所以实际的注释值在数量上并没有意义。
但是,可以比较每列中的缩放值。你可以很直观地感受到每个变异集群的相对属性。如果科学家要研发一种疫苗,则应该解决virii的主要病毒群。
下一步是运用PCA将集群可视化。
运用PCA将集群可视化
PCA是一种降维方法,选择多维空间中的正交向量来表示轴,这样一来就保留了大部分信息(方差)。
使用流行的Python库sklearn,可以用两行代码实现PCA。首先,我们可以检查解释的方差比。这是原始数据集中保留的统计信息的百分比。在这种情况下,解释的方差比是0.9838548580740327,这简直是天文数字!
可以确信的是,我们从PCA中得到的任何分析都将是真实的数据。
每个新特征(主成分)都是几个其他列的线性组合。我们可以用热图直观地看到一个列对两个主要组件中的任何一个都很重要。

重要的是要理解第一个组件中的高值意味着什么——在这种情况下,其特征是对齐长度较长,也就是更接近原始病毒,而组件2的特征主要是对齐长度较短,也就是突变后更远离原始值,这也反映在bit score较大的差异上。

很明显,病毒突变有5条主线。我们可以从中得到很多信息。
其中有四个病毒突变位于第一主成分的左侧,一个位于右侧。第一主成分的特征是高对齐长度。这表示第一主成分的较高值对应较高的对齐长度(更接近原始病毒)。
因此,组件1的较低值与原始病毒的基因差距较大。大多数病毒集群与原始病毒有很大不同。因此,试图制造疫苗的科学家应该意识到这种病毒会发生大规模变异。
通过使用K-Means和PCA,我们可以识别出五种主要的新冠病毒变异集群,研制疫苗的科学家可以运用这些集群中心获得的每种集群特征。通过PCA,我们可以在两个维度上看到这些集群中心,并且发现冠状病毒具有非常高的突变率。
这可能就是新冠病毒如此致命的原因。

Recommend
About Joyk
Aggregate valuable and interesting links.
Joyk means Joy of geeK