

哥大团队发布首个AlphaFold 2完整复刻版模型,在推理速度、内存占用等方面更具优势
source link: https://www.mittrchina.com/news/detail/10787
Go to the source link to view the article. You can view the picture content, updated content and better typesetting reading experience. If the link is broken, please click the button below to view the snapshot at that time.
麻省理工科技评论-哥大团队发布首个AlphaFold 2完整复刻版模型,在推理速度、内存占用等方面更具优势
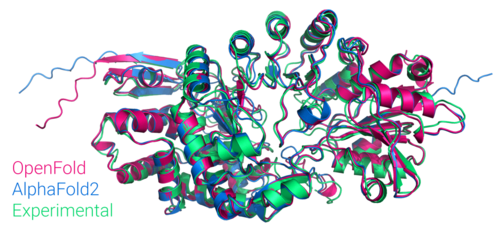
近日,有专家复刻了 AlphaFold 2 模型,以让大众能更容易使用其功能。
该复刻版本名为 OpenFold,采用 PyTorch 框架。据 PyTorch 官网:“PyTorch 是一个开源机器学习框架,可加速从研究原型到生产部署的路径。”
AlphaFold 2 是由 AI 公司 DeepMind 开发的深度学习系统,最为人知的是被用于预测蛋白质结构。这可以看作 AI 用于推动科学研究的一个里程碑事件。
对于蛋白质结构的预测有望在生命科学领域产生各种各样的好处,比如加速先进药物的发现和更好地理解疾病。AlphaFold 2 在 2020 年 11 月的蛋白质结构预测技术关键评估(CASP,Critical Assessment of protein Structure Prediction)比赛中总体排名第一(其第一代版本在 2018 年 12 月第 13 届 CASP 大赛中同样获得第一)。这被认为是计算生物学的重大成就,也是朝着数十年来生物学的重大挑战取得的巨大进展。
2021 年 7 月,用 AlphaFold 2 预测蛋白质结构的相关论文发表在 Nature,题为《用 AlphaFold 可以高精度地预测蛋白质结构》(Highly accurate protein structure prediction with AlphaFold)。同时,DeepMind 还向公众公布了蛋白质组数据库,并将 AlphaFold 2 开源。
但 AlphaFold 2 的实际部署比较困难,硬件要求、空间占用、下载时间长等方面对普通开发者不够友好。因此,相关专家和开源社区都在致力于打造一个方便使用的 AlphaFold 2 版本。
本次哥伦比亚大学系统生物学系助理教授穆罕默德·库莱希(Mohammed AlQuraishi)表示, 他们构建的可训练 OpenFold 模型是 AlphaFold 2 的第一个完整公开复制品,并已在 GitHub 开源。
开发人员表示:“OpenFold 再现了(几乎)原始开源推理代码(v2.0.1)的所有功能。我们已经公开发布了模型权重和训练数据,大约 40 万个 MSA(测量系统分析,Measurement Systems Analysis)和 PDB70 文件。”
据了解,模型权重可通过 GitHub 存储库中的脚本获得,而 MSA 则由 AWS(Amazon Web Services,亚马逊网络服务)上的开放数据注册表(RODA) 托管。OpenFold 的所有代码都基于 PyTorch 环境,而 AlphaFold 2 是为 JAX 工作流开发的。此外,除了 AlphaFold 的官方参数,OpenFold 支持使用开发人员自己的 Colab notebook 进行推理。
然后,值得一提的是,相比原版 AlphaFold 2,OpenFold 甚至在推理速度、内存占用方面要更具优势。比如超长链的推理、更快的短链推理(速度约为 AlphaFold2 的两倍)、高效的对齐脚本等方面。
研究者表示,在 40GB A100 上最多可以得到约 4600 个氨基酸残基的序列结构,并可以进一步优化。凭借新的可自定义 CUDA 注意内核,占用的 GPU 内存比 FastFold 内核少近四倍。
根据 CAMEO 的验证集上的 GDT_TS 分数,将 OpenFold 和 AlphaFold 2 比较,可以从下面的散点图看到两者的准确度十分接近。甚至平均而言 OpenFold 稍好。研究人员解释说,可能是由于他们的训练集更大。
据了解,OpenFold 在 A100 GPU 上训练了大约 10 万个计算小时,但 90% 的最终精度仅在前 3000 个计算小时内就达到了。研究人员表示,在最初的快速增长之后,精度增益显着减慢,尽管它仍然逐渐攀升。这对训练 OpenFold 和 AlphaFold 2 变体具有重要意义。
他还提到,本次采用的 GPU 为英伟达发布的 A100,后续希望能在较低端 GPU 上进行训练。目前他们有一个 AlphaFold-Gap 选项,应该很快就会启动并运行多聚体版本(使用 AF2-multimer 权重)。
研究者还表示,即将发表相关的预印本文章,其中包含在培训和研究的大量细节。并说:“我们的 OpenFold 努力远未结束。事实上,这只是一个开始。请继续关注后续发布的激动人心的消息。”
最后,蛋白质折叠是科学家几十年来一直面临的一个问题。据维基百科:“蛋白质折叠是蛋白质链被翻译成其天然三维结构的物理过程,通常是蛋白质变得具有生物功能的‘折叠’构象。”
目前,对其的研究一般通过“X射线晶体学、冷冻电子显微镜和核磁共振”等技术进行实验测定,然而,这些技术的使用在时间和成本方面都耗费较大。
如果能够仅从氨基酸序列中预测蛋白质结构,将大大有助于推进科学研究,可能会导致医学和生物学理解的全面快速突破。
蛋白质折叠涉及了决定折叠稳定结构的原子间力的热力学、蛋白质极快地达到其最终折叠状态的机制和途径,以及如何从其氨基酸序列中预测蛋白质的天然结构等极其复杂问题。
之前,研究人员也应用了许多计算方法来解决蛋白质结构预测的问题,但除了小的简单蛋白质外,它们的准确性并不接近实验技术,从而限制了其价值。OpenFold 与 AlphaFold 2 等 AI 模型预计会在蛋白质折叠问题上发挥越来越重要的作用。
-End-
参考:
https://twitter.com/MoAlQuraishi/status/1459188604723351552
https://github.com/aqlaboratory/openfold#readme
https://en.wikipedia.org/wiki/AlphaFold
Recommend
About Joyk
Aggregate valuable and interesting links.
Joyk means Joy of geeK